



Структурный тип PbZr3O4F6 (a ≈ 2a (флюорит), Z = 8, пр. гр. симметрии Fm-3m, позиции атомов Pb(8c), Zr(24e), O(32f), F(48i)) является производным от структурного типа KY3F10, который относится к группе A2B6X20-22 семейства флюоритоподобных фаз {A8–xB6CyXn+2(y–x)}m [1]. Особенностью данного семейства является возможность изо- и гетеровалентных замещений в катионной и анионной подрешетках, что открывает перспективы поиска новых структур, способных служить основой для создания материалов с заданными физико-химическими свойствами. Моделирование кристаллических структур позволяет существенно уменьшить объем экспериментальных исследований с целью поиска новых перспективных материалов, так как ограничивает диапазон возможных вариантов, как по качественному, так и по количественному составу.
В настоящей работе рассмотрены варианты изовалентных замещений катионов Zr4+ в структуре PbZr3O4F6 на катионы Hf4+, Ce4+, U4+, Np4+, Th4+, которые по кристаллохимическим критериям способны образовывать структуры, относящиеся к семейству флюоритоподобных фаз {A8–xB6CyXn+2(y–x)}m. Для оценки возможности образования новых структур использована концепция сумм валентностей связей (bond valence sums, BVS), широко применяемая в современной кристаллохимии неорганических ионных соединений [2]. Согласно данной концепции сумма валентностей связей каждого иона структуры равна абсолютному значению формального заряда данного иона (степени окисления):
|Z|=∑s.
Результаты моделирования базовой кристаллической структуры PbZr3O4F6 свидетельствуют о корректности использования данной концепции [3]. Относительные отклонения теоретически рассчитанных структурных параметров от экспериментальных не превышали 2%.
При расчетах в качестве стартовой модели использовались координаты атомов структуры PbZr3O4F6. При моделировании структур минимизировался предложенный в [4] функционал Ф, учитывающий не только взаимодействия катион-анион, но и межанионное отталкивание:
Ф=∑(ΔZi)2+∑[B/(dX-X)12]/2,
где ΔZi – разность между табличным и рассчитанным зарядом иона, dX-X – расстояние анион-анион, B – эмпирическая константа. Наши предыдущие исследования показали, что расстояниями катион-катион можно пренебречь, так как их учет не вносит вклад в окончательные результаты.
Расчет валентностей связей осуществлялся по экспоненциальной зависимости [2]:
s = exp((R0 – d)/b),
где s – валентность связи катион-анион, R0 – эмпирический параметр, характеризующий данную связь, d – межатомное расстояние, b – эмпирическая константа, равная 0.037 нм. Использовались значения параметров R0, приведенные в [5] (R0(Pb-O) = 0.2112 нм, R0(Pb-F) = 0.203 нм). Корректность полученных моделей структур оценивалась по глобальному индексу нестабильности (global instability index) GII [2], значения которого меньшие чем 0.1 свидетельствуют о стабильности кристаллической структуры:
GII = [∑(d2/N)]1/2,
где d – разность между табличным и рассчитанным зарядом для N ионов в независимой части элементарной ячейки.
Для структур PbM3O4F6 (M = Hf4+, Ce4+, U4+, Np4+, Th4+) теоретически рассчитанные абсолютные значения зарядов ионов практически совпадают с общепринятыми значениями. Глобальный индекс нестабильности GII имеет небольшое значение менее 0.01 (таблица), что указывает на большую вероятность возможности их существования.
Результаты моделирования кристаллических структур PbM3O4F6.
|
Структура |
R0(M-O), нм |
R0(M-F), нм |
Параметр |
Координаты атомов |
Индекс |
||
|
элементарной ячейки а, нм |
x(M) |
x(O) |
y(F) |
GII |
|||
|
PbHf3O4F6 |
0.1923 |
0.185 |
1.0894 |
0.2248 |
0.1141 |
0.1651 |
0.0012 |
|
PbCe3O4F6 |
0.2028 |
0.1995 |
1.1336 |
0.2191 |
0.1225 |
0.1670 |
0.0002 |
|
PbU3O4F6 |
0.2112 |
0.2038 |
1.1575 |
0.2202 |
0.1266 |
0.1649 |
0.0001 |
|
PbNp3O4F6 |
0.218 |
0.202 |
1.1668 |
0.2275 |
0.1277 |
0.1679 |
0.0001 |
|
PbTh3O4F6 |
0.2167 |
0.2068 |
1.1724 |
0.2219 |
0.1285 |
0.1678 |
0.0001 |
Полученные межатомные расстояния для структур PbM3O4F6 находятся в пределах, характерных для кристаллических структур, содержащих данные ионы. Рассчитанные рентгенограммы модельных структур четко указывают на их принадлежность к структурному типу PbZr3O4F6 (рисунок) и могут быть использованы при идентификации новых фаз в дальнейших исследованиях.
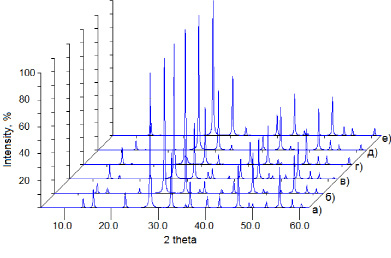
Рассчитанные рентгенограммы структур PbZr3O4F6 (а), PbHf3O4F6 (б), PbCe3O4F6 (в), PbU3O4F6 (г), PbNp3O4F6 (д) и PbTh3O4F6 (е). Излучение CuKα, λ = 0.154056 нм
Таким образом, проведенное моделирование кристаллических структур PbM3O4F6 (M = Hf4+, Ce4+, U4+, Np4+, Th4+), относящихся к структурному типу PbZr3O4F6, свидетельствует о возможности их существования.