



В работах [1, 2] описан экспериментальный подход к триазолоксидам. Интерес к соединениям подобного строения обусловлен тем, что некоторые из них обладают противоопухолевой активностью. учитывая это, а также недостаточно глубокую информацию о свойствах конденсированных триазолоксидов мы рассмотрели экспериментальные и квантово-химические особенности их образования, как было показано ранее [2]. Известно, что в результате реакции аминирования нафтохинонов (I) образуются продукты (II), которые в дальнейшем переходят в нафтатриазолоксиды.
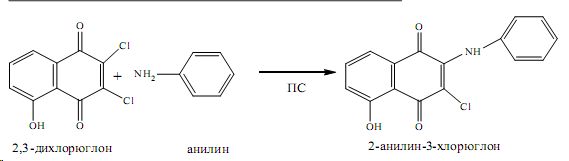
Первая стадия этой реакции представлена на схеме.
Целью настоящей работы явилось исследование методом функционала плотности механизма первой стадии реакции конденсирования дихлорюглона с анилином и сравнение с экспериментальными данными. Для всех расчетов был использован полноэлектронный базисный набор 6-31G(d) с функционалом плотности B3LYP. Данный метод расчета широко используется для анализа термодинамических параметров органических соединений.
Все расчеты были проведены с использованием стандартного пакета программ GAUSSIAN´03. Для проведения теоретических исследований был использован квантово-химический метод функционала плотности (DFT, Density Functional Theory). расчеты проводили гибридным методом функционала плотности B3LYP, с обменным функционалом Беке (В3) [3] и корреляционным функционалом ли, Янга и Пара (LYP) [4]. Для всех атомов использовался полноэлектронный базисный набор 6-31G(d). геометрии всех рассчитанных молекул были полностью оптимизированы, отсутствие мнимых частот колебании подтверждало их стационарный характер. Оптимизация переходных состояний проведена с использованием метода STQN, переходные состояния имели только одну мнимую частоту. расчеты в растворе этанола проведены теми же методами с использованием модели поляризованного континуума (PCM). Энергии рассчитанных соединений скорректированы с учетом нулевой колебательной энергии (ZPVE) и приведены к стандартным условиям (298.15 К, 1 атм.) с использованием термической поправки к энтальпии и свободной энергии.
Точность любых квантово-химических расчетов определяется согласием экспериментальной и рассчитанной геометрии молекул. Сравнение вычисленных нами длин связей (R) и валентных углов (ω) с экспериментальными данными показывает, что рассчитанные длины связей в основном занижены, а валентные углы завышены. Однако проведенная статистическая обработка результатов приводит к следующим корреляционным соотношениям между рассчитанными и экспериментальными длинами связей и валентными углами [5] для некоторых изученных молекул:
Rэксп. = -0.04 + 1.03 Rрасч.
(r = 0.996; s = 0.02; n = 22) (1)
ωэксп. = -17,9 + 1,14 ωрасч.
(r = 0.982; s = 1.5; n = 32) (2)
В этих и следующих корреляционных уравнениях r - коэффициент корреляции, s - стандартное отклонение, и n - число соединений, входящих в корреляцию.
Для подтверждения правильности проведенных нами расчетов мы провели сопоставление рассчитанных и экспериментальных [6] длин волн уФ-спектров (λ) и спектров ЯМр 1Н и 13С (δ) некоторых хиноидных соединений. Корреляционные уравнения (3-5) показывают, что проведенные нами расчеты позволяют оценивать спектральные параметры с достаточной степенью точности, что свидетельствует о том, что рассчитанные структуры молекул можно использовать для выявления термодинамических параметров реакций аминирования.
λ эксп. = -16 + 1.06 λ расч.
(r=0.998; s=7; n=22) (3)
δэксп. 1Н = 1,23 + 0,83 δрасч. 1Н
(r=0.995; s=0,2; n=19) (4)
δэксп. 13С = 4,7 + 0,92 δрасч. 13С
(r=0.996; s=2,2; n=8) (5)
Из таблицы 1 видно, что все приведенные реакции термодинамически выгодны как в газовой фазе, так и в растворителе. Однако в растворителе свободная энергия гиббса имеет примерно в два раза большее значение вследствие сольватации реагентов этиловым спиртом.
Для выяснения возможного механизма реакции нами были рассчитаны переходные состояния для ряда реакций аминирования и из полученных данных были вычислены
Таблица 1. Термодинамические характеристики реагентов, продуктов и переходных состояний (кДж/моль)
|
Реагент |
Ариламин |
ΔH |
ΔG |
ΔG≠ |
E |
|
|
Газ. фаза |
EtOH |
|||||
|
2,3-дихлор-1,4нафтохинон |
анилин |
-46 |
-36 |
-45 |
80 |
94 |
|
п-толуидин |
-46 |
-42 |
-47 |
78 |
91 |
|
|
м-толуидин |
-45 |
-41 |
-45 |
74 |
94 |
|
|
анизидин |
-60 |
-45 |
-59 |
64 |
76 |
|
|
п-хлоранилин |
-44 |
-34 |
-40 |
89 |
100 |
|
|
2,3-дихлорюглон |
анилин |
-51 |
-41 |
-47 |
75 |
86 |
|
п-толуидин |
-51 |
-47 |
-43 |
73 |
85 |
|
|
м-толуидин |
-49 |
-45 |
-41 |
77 |
87 |
|
|
анизидин |
-65 |
-50 |
-55 |
98 |
68 |
|
|
п-хлоранилин |
-48 |
-37 |
-36 |
85 |
93 |
|
|
2,3-дихлорнафтазарин |
анилин |
-48 |
-38 |
-445 |
80 |
93 |
энергии активаций данных реакций в газовой фазе по уравнению:
Ea = ΔH≠ +nRT, (6)
где Еа - энергия активации, ΔН≠ - энтальпия активации, оцененная по энтальпии реагента и переходного состояния, n - порядок реакции, R - газовая постоянная, Т - абсолютная температура. Однако известно, что представленные реакции в реальности проходят в растворах [2]. Мы рассчитали энергию активации в растворе этилового спирта как разность свободных энергий гиббса в растворе реагента и переходного состояния (ΔG≠).
Результаты также представлены в табл. 1. рассчитанные значения энергии активации меньше в растворе, чем в газовой фазе и близки к известным экспериментальным значениям SNAr реакций [7]. По результатам расчета во всех случаях переходные состояния представляют собой интермедиаты, в которых происходит отщепление атома хлора от нафтохинона и образование слабой связи между атомами углерода нафтохинона и азота аминогруппы (1,8Å).
Таким образом, полученные результаты позволяют доказать, что реакция аминирования нафтохинонов проходит через рассчитанное нами переходное состояние.
Список литературы
- Churakov A.M., Ioffe S.L., Strelenko Yu.A., Tartakovsky V.A. // Tetrahedron Letters, 1996, V. 37. р. 8577-8583.
- Радаева Н.Ю., Долгушина Л.В., Сакилиди В.Т., Горностаев Л.М. // Журнал органической химии, 2005, Т. 41. С. 926-930.
- Becke A.D. // J. Chem. Phys., 1993, V. 98. р. 5648-5655.
- Handy N.C., Cohen A.J. // Mol. Phys., 2001, V. 99. р. 403-410.
- Standard Reference Database 17, Version 7.0 (Web Version), Release 1.4.2
- NIST Chemistry WebBook http://webbook.nist.gov/chemistry
- Computational Organic Chemistry: Ed. S. M. Bachrach, John Wiley&Sons. Inc. 2007, 478 p.
Библиографическая ссылка
М. Н. Зверева, О.Х. Полещук, А. Г. Яркова, Л. Н. Долгушина, Л .М. Горностаев ИССЛЕДОВАНИЕ МЕХАНИЗМА РЕАКЦИИ АМИНИРОВАНИЯ МЕТОДОМ ТЕОРИИ ФУНКЦИОНАЛА ПЛОТНОСТИ // Современные наукоемкие технологии. 2010. № 6. С. 7-10;URL: https://top-technologies.ru/ru/article/view?id=24846 (дата обращения: 03.07.2026).