



Решения, основанные на технологиях математического моделирования, машинного и глубокого обучения, широко используются во всех сферах нашей жизни, и такие области знания, как биология, химия и медицина, здесь не исключение. Например, моделирование различных терапевтических воздействий на пациента [1–3] является важным шагом на пути к персонализированной медицине. Внедрение указанных технологий в медицину также заключается в диагностике заболеваний при помощи технологий компьютерного зрения [4–6], мониторинга состояния пациентов, предсказания течения болезней, а особый класс моделей – генеративные модели [7] – позволяют создавать новые объекты. В рамках биологии, химии и хемоинформатики таковыми новыми объектами и структурами являются молекулярные соединения, в частности лекарственные. В данной статье будет рассмотрена тема генерации лекарственных молекулярных соединений при помощи нейронных сетей, которые будут обучены на специальном наборе данных, представляющих из себя набор известных химических молекул.
Следует отметить, что открытие и разработка нового лекарственного средства – это чрезвычайно длительный, дорогостоящий, сложный и неэффективный процесс, который занимает в среднем 10–15 лет. Несмотря на достижения в области технологий и очень хорошее понимание биологических систем, в последние два десятилетия в фармацевтической промышленности наблюдается все большее снижение производительности исследований и разработок из-за растущих затрат, в то время как абсолютное число вновь одобренных лекарств постоянно сокращается из-за постоянно растущих регуляторных препятствий и возрастающих трудностей в поиске следующего препарата. Таким образом, процесс создания лекарственного препарата становится дорогостоящим и трудоемким [8], а большинство новых одобренных лекарств – низкомолекулярные препараты.
С другой стороны, успехи генеративных моделей в компьютерном зрении и обработке текстов позволяют рассчитывать на то, что их применение в химии и биологии будет способствовать ускорению процесса разработки новых лекарственных соединений. Например, трансформерная модель AlphaFold2 [9] значительно увеличила качество предсказания протеиновых структур по сравнению с известными ранее моделями. Однако такие модели имеют огромное количество параметров, что делает затруднительным понимание их работы и возможности их обучения на стандартных средствах вычислительной техники. Другие генеративные модели, такие как вариационные автоэнкодеры (VAEs) [10], генеративные состязательные сети (GANs) [7] и рекуррентные нейронные сети (RNN) [11], специально разработаны для изучения скрытых представлений молекул и генерации большого количества кандидатов на лекарства для дальнейшего скрининга.
Нейронные сети широко используются для создания миллионов de novo молекул в известном химическом пространстве. Эти глубокие генеративные модели обычно настраиваются с помощью LSTM или GRU, которые обучаются на специальном представлении молекул SMILES – Simplified Molecular Input Line Entry System (упрощенная система строкового представления молекулярных соединений). В исследовании [12] авторы представляют модель нейронной сети, Generative Examination Networks, основанной на двунаправленной RNN с конкатенированные подмодели для обучения и генерации молекулярных строк SMILES.
Таким образом, анализ литературы показывает, что основными генеративными алгоритмами являются модели GAN и VAE. Однако таким алгоритмам, как сэмплирование с температурой, top-k sampling [13], уделяется недостаточное внимание. В данной статье было предпринято решение воспользоваться именно такими генеративными алгоритмами и сравнить их с алгоритмами VAE и GAN.
Основными целями исследования являются:
1) моделирование лекарственных молекулярных соединений на базе искусственных нейронных сетей и проверка адекватности генерируемых моделей по метрике qed;
2) показать, что с задачей генерации потенциальных лекарственных молекулярных соединений наравне с моделями gan, vae справляются генеративные алгоритмы сэмплирования.
Материалы и методы исследования
Задача генерации лекарственных молекулярных соединений при помощи строк SMILES в терминах нейронной сети является задачей обработки естественного языка (Natural Language Processing, NLP), в которой химическое пространство молекул, кодированных SMILES строкой, является языковой моделью. То есть для решения данной задачи требуется решить задачу NLP: генерация нового текста.
В данной работе в качестве набора данных будет использоваться датасет, состоящий из 250 000 химических молекул, которые кодированы по специальным правилам. Такие закодированные молекулы называются SMILES. SMILES – это популярный метод описания молекул с помощью текстовых строк. Такое представление описывает атомы и связи молекулы одновременно точно и достаточно интуитивно понятно. Например, строка «OCCc1c(C)[n+](cs1)Cc2cnc(C)nc2N» описывает важный питательный элемент тиамин, также известный как витамин B1.
В качестве основных генеративных алгоритмов использовались следующие: сэмплирование с температурой; Top-K Sampling (сэмплирование K-верхних); жадный поиск (Greedy search); VAE – вариационный автокодировщик.
Рассмотрим сэмплирование с температурой [14]. Само по себе случайное сэмплирование потенциально может сгенерировать совершенно произвольное слово. Чтобы избежать данного явления, вводится понятие “temperature” (t), для увеличения вероятности получения наиболее вероятных слов. Обычно берётся диапазон 0 < t ≤ 1. Условная вероятность следующего состояния описывается выражением
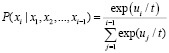 , (1)
, (1)
где u – вектор, содержащий значения каждого токена в словаре.
Генерация следующего слова будет производиться с распределением p’=softmax(log(p)/t). Тогда при t = 1 получаем p’ = p. При больших t сэмплирование происходит равновероятно. Однако при малых t всегда выбирается самый вероятный токен. То есть семплирование с температурой – это общий вид разных видов семплирования, в разной степени учитывающих предсказания модели. Это нужно, чтобы лавировать между уверенностью модели и разнообразием. Можно поднимать температуру, чтобы генерировать более разнообразные тексты, или опускать её, чтобы генерировать тексты, в которых модель в среднем более уверена.
Fan и соавт. [15] в 2018 г. представили простую, но очень мощную модель сэмплирования, называемую top-K sampling. В выборке Top-K фильтруются K наиболее вероятных следующих слов, и сумма вероятности перераспределяется только между этими K-следующими словами.
Жадный поиск (Greedy search) выбирает токен с наибольшей вероятностью в качестве следующего слова
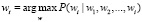
на каждом временном шаге t.
Последним предложенным решением стало создание модели вариационного автокодировщика (VAE). Данная модель была предложена в 2013 г. в статье [16]. Данная модель учится отображать некоторый объект в заданное скрытое пространство (latent space) и обратно. Ключевое отличие VAE от обычного автокодировщика заключается в наличии вариационного вывода. Данный метод используется для аппроксимации распределений, который использует процесс оптимизации по параметрам, чтобы найти наилучшее приближение среди данного семейства распределений. Структурно модель VAE состоит из следующих категорий: кодировщик, скрытое пространство, декодировщик. Модель VAE обладает уникальным свойством: скрытое пространство является непрерывным. Данное свойство помогает выполнять случайные преобразования и интерполяцию. Непрерывность достигается тем, что на выходе из кодировщика появляется два вектора: вектор средних значений и вектор стандартных отклонений.
Вариационные автокодировщики не лишены проблем и минусов. Первой является то, что для обучения двух нейронных сетей с помощью алгоритма обратного распространения ошибок нужно контролировать все этапы обучения распространения ошибки. Поскольку декодер не является детерминированным (оценка его вывода требует оценки по многомерному гауссову распределению). Второй проблемой является требовательность к ресурсам для обучения. Третьей проблемой вариационных автокодировщиков является чувствительность к наборам данных: если набор данных не идентифицирован должным образом, VAE не сможет изучить какое-либо соответствующее распределение вероятностей в наборе данных и впоследствии не сможет сгенерировать новые объекты на должном уровне.
В основе таких моделей лежит блок GRU, представленный в 2014 г. в статье [17]. По эффективности и по качеству обучения данный вид нейронной сети схож с известной LSTM, однако из-за того, что GRU имеет на один “gate” меньше, данный блок имеет меньше параметров и потенциально может быстрее обучаться и сходиться. Представим архитектуру GRU с помощью математической модели
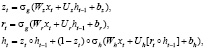
(2)
где ⸰ – это произведение Адамара; σg и σh – это две функции активации на основе сигмоиды и гиперболического тангенса соответственно; xt – это входной вектор; ht – это выходной вектор; zt – это вектор вентиля обновления; rt – это вектор вентиля сброса; W,U,b – это матрицы переменных и вектор свободных весов.
В качестве метрики будем использовать специальную химическую метрику QED [18]. В некоторых недавних публикациях о генеративных моделях для определения пригодности молекулы используют вычисляемые молекулярные свойства. Методика QED сравнивает распределение набора свойств, рассчитанных для молекулы, с распределениями тех же свойств в продаваемых лекарствах. Показатель совпадения варьируется от 0 до 1. При этом молекулы с показателем около 1 считаются наиболее похожими на лекарства. В качестве оценки мы рассчитаем QED для сгенерированных молекул и отбросим те, у которых показатели меньше 0,5. Данная метрика вычисляется в соответствии с выражением
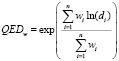 . (3)
. (3)
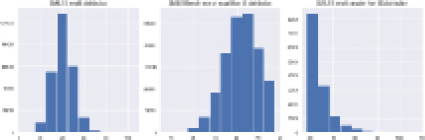
Рис. 1. Распределение атомов в молекулах
В данном уравнении d – это индивидуальная функция желательности, w – вес, применяемый к каждой функции, а n – количество молекулярных дескрипторов. Обычно функции желательности определяются произвольно. Как правило, это убывающие или возрастающие монотонные функции или функции «горба» в определенных диапазонах параметров и точках перегиба. Оптимальный набор весов – это тот, который максимизирует информационное содержание, которое может быть измерено путем вычисления энтропии Шеннона.
Результаты исследования и их обсуждение
Данные были взяты из открытых источников [19] и насчитывают порядка 250 000 молекулярных соединений, кодированных в SMILE структуру. Распределения атомов представлены на рис. 1.
Как можно видеть, больше всего молекул имеют в своем составе от 30 до 50 молекул, и лишь незначительная часть имеет больше 90. Максимальное же количество атомов в молекуле – 110.
Согласно рис. 2, в данном наборе данных можно заметить, что большая часть молекул имеют QED около 0,8, что говорит о достаточной схожести с лекарственными молекулами. Однако следует заметить, что лекарством может являться и молекулярное соединение с достаточно низким QED.
Обучение модели исследования V-GRU на основе GRU проводилось на полном наборе данных (250 000 молекул) с разбивкой выборки обучения на тестовую и валидационную с соотношением 0,8:0,2. Данная модель имеет 6 слоев GRU, 2 полносвязных слоя. Общее число параметров – 2 369 839. Процесс обучения представлен на рис. 3. Можно заметить, что данная нейронная сеть достаточно хорошо обучается на тренировочном наборе данных. Функция потерь уменьшается, а точность увеличивается. Эффекта переобучения нет.
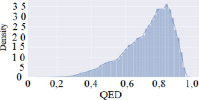
Рис. 2. Распределение свойства QED в наборе данных
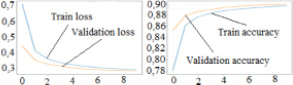
Рис. 3. Процесс обучения модели V-GRU
|
а) сэмплирование с температурой |
б) top-k sampling |
|
в) Жадный поиск |
г) V-VAE |
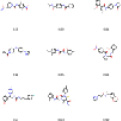
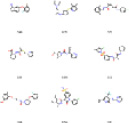
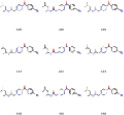

Рис. 4. Примеры генерируемых SMILES
В связи с ограниченностью ресурсов, обучение V-VAE проводилось не на полном наборе данных. Модель имеет слои энкодера и декодера. Всего 864 932 параметра. Для обучения были взяты 75000 молекул.
Результаты генерации представлены на рис. 4.
Таким образом, алгоритм жадного поиска не справляется с данной задачей, генерируя однотипные структуры.
Заключение
В данной работе мы предложили четыре алгоритма для создания потенциальных лекарственных молекулярных соединений.
В результате проделанной работы было сгенерировано некоторое множество лекарственных молекулярных соединений, которые были оценены метрикой QED. Данные соединения являются кандидатами на лекарства. Оставшаяся работа по проверке данных соединений на эффективность и безопасность входит в компетенцию научных сотрудников лабораторий фармацевтических компаний.
Таким образом, основная гипотеза данного исследования выполняется: мы показали, что с задачей генерации лекарственных молекулярных соединений наравне с моделями GAN, VAE справляются генеративные алгоритмы сэмплирования.
В качестве дальнейших направлений исследований можно рассмотреть применение более инновационных алгоритмов с целью достижения желаемых результатов: GAN, квантовый GAN, WGAN, CVAE или другие алгоритмы сэмплирования: beam search или nucleus sampling. Также в спецификации модели можно учесть уязвимый белок болезни, на который должно воздействовать желаемое лекарственное соединение.
Библиографическая ссылка
Веселов Д.И., Андриянов Н.А. ПРИМЕНЕНИЕ ГЕНЕРАТИВНЫХ МОДЕЛЕЙ ДЛЯ ПРЕДСКАЗАНИЯ ЛЕКАРСТВЕННЫХ МОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ // Современные наукоемкие технологии. 2022. № 3. С. 16-21;URL: https://top-technologies.ru/ru/article/view?id=39067 (дата обращения: 01.08.2026).
DOI: https://doi.org/10.17513/snt.39067