



При конструировании эффективных физиологически активных веществ прежде всего необходимо учитывать молекулярную структуру исходных соединений и их физико-химические свойства, влияющие на транспорт веществ в организме и их взаимодействие с биологическими мишенями. Исключительно важная роль пространственной структуры обуславливает создание и использование различных дескрипторов, описывающих межмолекулярные взаимодействия в трехмерном пространстве. Ранее нами [2-4] были предложены дескрипторы, рассчитываемые из спектра межатомных расстояний.
Описание дескрипторов
При использовании спектров межатомных расстояний молекулярная структура представляется в виде кривой функции радиального рассеяния, которая легко может быть вычислена для молекулы любой сложности, если известны ее декартовы координаты. Аналогичный аппарат применяется при изучении строения молекул методом газовой электронографии [5].
Функция радиального распределения, M(s) в приближении сферической симметрии атомов и отсутствии внутримолекулярных колебаний представляет собой спектр межатомных расстояний, интенсивность которого определяется видом атомов, находящихся на данном расстоянии друг от друга, и динамикой изменения этого расстояния в процессе внутримолекулярных колебаний:
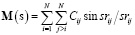 (1)
(1)
где s – угловой параметр (s= (4p/l)sin Q/2), l – длина волны электронов, Q – угол рассеяния, rij – межатомное расстояние в молекуле, Cij – коэффициент, характеризующий рассеивающую способность данной пары атомов.
Можно сказать, что функция радиального распределения, получаемая в процессе электронографического эксперимента, это то, что «видит» электрон, который взаимодействует с молекулой в акте рассеяния.
В публикации [5] эта аналогия была впервые продолжена на случай взаимодействия рецептора с молекулами введенного в организм вещества. Как известно, взаимодействие рецептора происходит не со всей молекулой реагента, а с частями молекулы, комплементарными активным центрам рецептора. При этом определяющими факторами взаимодействия являются расстояния между активными центрами и способность к межмолекулярным взаимодействиям атомов, находящихся на этих расстояниях в молекулах реагента. Имеет место своеобразный процесс рассеяния молекул реагента на активных центрах рецептора, т.е. рецептор ”видит” молекулу в образе функции радиального распределения. При таком подходе существует возможность модифицировать функцию радиального распределения таким образом, чтобы количественно учесть способность атомов реагента к различным межмолекулярным взаимодействиям. В публикациях [5, 6] было высказано предложение учитывать вместо типа атомов их донорно-акцепторную способность. Таким образом, вместо интенсивности рассеяния M(s) (1) в спектре рассчитываются энергии следующих взаимодействий:
1. стерическое взаимодействие атомов (Van der Waals)
2. взаимодействие положительно заряженных атомов,
3. взаимодействие отрицательно заряженных атомов,
4. взаимодействий положительно и отрицательно заряженных атомов,
5. взаимодействие атомов – доноров водородной связи,
6. взаимодействий атомов – акцепторов водородной связи,
7. взаимодействие атомов – доноров с атомами – акцепторами водородной связи.
Реально спектр представлен в виде суммы пиков (рисунок), представленных функцией Гаусса, в которых в качестве интенсивности фигурирует энергия одного из указанных выше взаимодействий, в качестве полуширины фигурируют смещения атомов в приближении теории малых колебаний, положение пика определяется расстоянием между соответствующими атомами.
Первая версия программы (Moltra-I) [2] является морально устаревшей и написана для Windows 3. Новая версия работает в качестве приложения СУБД CheD [1] и может использоваться как с 32 так и с 64 разрядными версиями Windows. Кроме этого введены новые функции, которые делают более удобными и наглядными процессы расчета дескрипторов и их визуализации.
Описание программы
Будучи приложением СУБД CheD [1] МОЛТРА обладает всеми ресурсами CheD по созданию и поддержке баз данных, в основе которых заложена 2D структура.
Ввод данных. Входными данными для работы МОЛТРА являются файлы в формате MOL2, содержащие, трехмерные координаты атомов. Эти файлы могут быть экспортированы одной из многих программ, оперирующих с трехмерными структурами, как экспериментальными так и расчетными.
Визуализация 3D структуры. Набор средств в окне структуры позволяет расположить молекулу наиболее удобным способом, чтобы сопоставить выделенный пик на спектре расстояний с парой атомов, которым этот пик соответствует. С другой стороны если выбрать пару атомов на структуре, то расстояние между ними отображается на спектре межатомных расстояний.
Работа со спектром межатомных расстояний. Для упрощения сложного спектра межатомных расстояний может быть использована система фильтров. Это позволяет отображать на спектре расстояния только для избранных атомов, например, доноры водорода NH, OH, атомы ароматических систем. Пики из спектра могут быть исключены и по признаку интенсивности, например, более 0.5.
Стандартные инструменты для работы со спектральной картиной, такие как протяжка (scroll) и растяжка (zoom), интегрирование а также указатели пиков и курсора активны в окне спектра. При выборе соответствующего пика на 3D структуре отображается пара атомов с наибольшей интенсивностью данного пика (если их несколько). Спектр может быть распечатан и экспортирован в графический формат.
Для сравнения могут быть отображены несколько спектров в базе в режимах:
а) Perspective – псевдо 3D пространство
б) Overlapped – спектры нескольких структур как есть
в) Columns – спектры нескольких структур со смещением по вертикали во избежание перекрывания
г) Average – среднеарифметический спектр для нескольких структур.
Расчет дескрипторов на основе спектра межатомных расстояний. В качестве дескрипторов спектра межатомных расстояний предлагаются интегралы спектра в определенном интервале, когда в качестве интенсивности используется энергетические свойства одного из взаимодействий (Van der Waals, кулоновские, донорно-акцепторные). Наличие выбранного взаимодействия на данном расстоянии и его количественная мера в виде интеграла может быть использована как дескриптор в процессах моделирования QSAR. Интегралы в спектре могут быть рассчитаны с заданным шагом и затем экспортированы в файлы общедоступных форматов (текст, exсel).
Расчет сходства спектров. Для поиска закономерностей в спектрах различных молекул очень полезна процедура расчета схожести спектров в заданном интервале.
Согласно статистическому критерию Танимото схожесть спектров может быть определена следующей формулой
Similarity(i,j)= S(i,j)/[S(i)+S(j)–S(i,j)] (2)
где S(i) – интеграл текущего спектра (i) в определенном интервале, S(j) – интеграл спектра сравнения (j) в том же интервале , S(i,j) – интеграл перекрывающейся части обоих спектров.
В результате работы процедуры расчета схожести спектров формируется таблица, в которой в первой строке прописана структура, с которой происходит сравнений (S(i,i)=1), далее прописываются структуры в порядке убывания схожести (S(i,j)). Таким образом в массиве данных могут быть определены молекулы с наиболее схожими спектрами. Результаты расчета могут быть экспортированы в текстовый файл и использованы в последующем моделировании.
Расчет спектров межатомных расстояний субстратов P-гликопротеина. Полезность МOLTRA для моделирования физиологической активности может быть продемонстрирована на примере поиска субстратов с белком P-гликопротеином, который ограничивает проницаемость веществ в мозг и играет ведущую роль в защите центральной нервной системы.
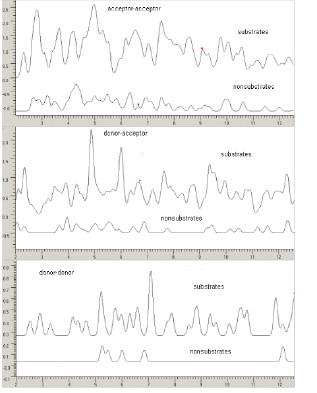
Фрагменты усредненных спектров межатомных взаимодействий для субстратов и несубстратов P-гликопротеина
В литературе [7] предполагается наличие двух характеристических расстояний 2.5±0.3Å (I) и 4.6±0.6Å (II) между донорами электронов в молекулах для образования субстратов с P-гликопротеином. Анализ спектров межатомных расстояний позволяет проверить наличие таких свойств и предположить наличие других характеристических расстояний для различных типов взаимодействий (H-донор « H-акцептор, H-акцептор « H-акцептор и H-донор « H-донор). На рис 1. представлены усредненные спектры для субстратов и несубстратов из массива 51 соединений [8] для различных типов взаимодействий. Структура рассчитана методом молекулярной механики ММ3 [32]. Спектры приведены в единой шкале интенсивности, которая представляет собой произведение H-донорных и Н-акцепторных факторов [9] для атомов находящихся друг от друга на рассматриваемом расстоянии. Усредненный спектр субстратов для взаимодействия типа акцептор-акцептор (оба атома электронодоноры) указывает на наличие характерных пиков близких к работе [7] (2.8Å и 4.9Å). В спектре субстратов для взаимодействия типа H-донор <-> H-акцептор присутствуют также два интенсивных пика в области 4.9Å и 6Å.
Таким образом, конкретное расположение и интенсивность пиков в указанных спектрах могут служить основанием для осторожных гипотез о возможной структуре комплексов между лигандами и P-гликопротеином. Дальнейшая проверка таких гипотез возможна при наличии точной информации о структуре таких комплексов с помощью рентгеновских и других современных методов.
Библиографическая ссылка
Трепалин С.В., Ярков А.В., Раздольский А.Н., Раевский О.А. MOLTRA-II, НОВЫЕ ВОЗМОЖНОСТИ ПРОГРАММЫ ДЛЯ РАСЧЕТА МОЛЕКУЛЯРНЫХ ДЕСКРИПТОРОВ НА ОСНОВЕ СПЕКТРА МЕЖАТОМНЫХ РАССТОЯНИЙ // Современные наукоемкие технологии. 2014. № 12-1. С. 33-36;URL: https://top-technologies.ru/ru/article/view?id=34805 (дата обращения: 10.06.2026).