


Scientific journal
Modern high technologies
ISSN 1812-7320
"Перечень" ВАК
ИФ РИНЦ = 1,279
EXPRESS ANALYSIS OF CONCENTRATION PARAFFIN AND POUR POINT BY NMR FUEL

В последнее десятилетие резко повысились требования к качеству дизельных (ДТ) и арктических топлив, в частности, к температуре застывания ТЗ. Поскольку ТЗ непосредственно зависит от концентрации высших алифатических предельных углеводородов (УВ) – парафинов (П), возросли требования к качеству депарафинизации (ДП) нефти и нефтепродуктов, в частности, ДТ и соответственно требования к экспресс-контролю параметров нефти и ДТ при процессах ДП. Кроме того, возникает необходимость принятия специальных мер по предупреждению накопления асфальто-смолисто-парафиновых отложений (АСПО) в скважинах и трубопроводах, что требует знание состава АСПО.
Современные методы определения параметров парафинов, асфальтенов и смол
Температура застывания ТЗ является одним из трудно определяемых параметров [1], поскольку процесс застывания в нефти и нефтепродуктах проходит в значительном временном и концентрационном интервале с выделением мелких кристалликов наиболее высокоплавкого и малого по содержанию парафина. На приборах регистрируется не начало фазового перехода, а промежуточный момент процесса, зависящий от чувствительности прибора, когда кристаллики П приобретают значительные размеры.
Известен ряд методов определения ТЗ: визуальный, ультразвуковой, вискозиметрический, фотометрический, объемный и калориметрический. Недостаток визуального – малый наблюдаемый объем и соответственно низкая представительность образца. При достаточно точном ультразвуковом методе при облучении изменяется сам процесс кристаллизации из-за формирования ультразвуком новых центров кристаллизации. Вискозиметрический метод недостаточно точный, поскольку требует значительного количества выделившегося П. Фотометрический метод позволяет определять ТЗ только прозрачных растворов. Авторы [1] предложили диэлькометрический метод, но не привели метрологических параметров метода.
В практике нефтехимических лабораторий используются методы определения параметров парафинов и асфальтенов в нефтях, стандартизованные по ГОСТ 11851 и ГОСТ 11858 соответственно. Первый метод заключается в предварительном удалении асфальтово-смолистых веществ (АСВ) из нефти, их экстракции и адсорбции и последующем выделении парафина смесью ацетона и толуола при Т = – 20 °C. Второй метод заключается в предварительном удалении АСВ вакуумной перегонкой с отбором фракций 250–550 °C и выделении парафина смесью спирта и эфира при Т = – 20 °C.
Разработана также методика определения массовых концентраций А, С и П в нефти в соответствии с ГОСТ 8.563-96, аттестованная Госстандартом РФ (M 01-12-81). В соответствии с ней определение А, С и П основано на применении стадий: осаждение А растворителем; выделение из деасфальтизированного остатка смолистых соединений методом комплексообразования с тетрахлоридом титана с последующим разложением комплекса и выделением смол; вымораживание парафина из деасфальтизированного и обессмоленного остатка нефти. Диапазон измерений: А – 0,3–15 %, С – 2,0–30 %, П – 2,0–15 %. Погрешность измерений: асфальтенов ± 0,60 %, смол ± 0,10 %, парафина ± 0,30 %.
Известен также способ [2] определения содержания парафинов и асфальтенов в нефти, заключающийся в отборе трех образцов сырой нефти, растворении двух в растворителе, удалении растворителя вместе с легкими фракциями. Для всех трех образцов методом ядерного магнитного резонанса (ЯМР) измеряют кривые спада свободной индукции и определяют отношение твердотельных фракций (ТФ) к водородсодержащим жидким фракциям. О концентрации П судят по содержанию ТФ в обработанном образце, из которого удалены А. О концентрации А судят по содержанию ТФ в другом образце с учетом установленной концентрации П.
Для определения содержания П также используется анализатор PRFC-A по ГОСТ 17789 – IP 459-1 – EN 12 606-1 (бывший DIN 52 015). Он включает дистилляционное и фильтрующее устройства, в состав которого входят: охлаждающая баня; специальные испытательные пробирки (NS 29/32) с предохранительными дистилляционными насадками; вакуумная колба; водоструйный насос; U-образный манометр.
В работе [3] делается вывод, что на настоящий момент ощущается потребность в экспресс-анализаторе, способном контролировать концентрацию парафина и температуры застывания и помутнения, не разделяя сырье на фазы и не используя движущихся деталей [3]. Особенно это актуально для нефтедобычи и переработки.
Наши исследования [4, 5] показывают, что из всех известных метод ЯМР способен одновременно контролировать ТЗ и ТМ концентраций П, А и С в твердой и жидкой фазах, а также такие важные характеристики водоорганических смесей, как концентрацию воды, 0дисперсность (распределение размеров капель воды), вязкость h20 и плотность r2 фаз и конечного продукта. ЯМР в портативном автономном и проточном варианте обладает возможностью экспресс-контроля в широком диапазоне, малой чувствительностью к примесям, достаточной точностью, возможностью управления и актуален для совершенствования процессов ДП.
Аппаратура ЯМР экспресс-анализа
Анализ рынка показывает, что таких ЯМР-анализаторов нет, и не только в РФ. Существующие анализаторы имеют высокие ошибки измерения и рассчитаны на однофазные, однородные среды. Требуется универсальный способ и устройство контроля в неконтактном и автоматическом экспресс-режиме. Этим условиям удовлетворяет метод ядерного (протонного) магнитного резонанса (ЯМР). Для реализации экспресс-анализа нами был разработан проточный ЯМР-анализатор [6] и проведены его испытания. Предложены методики экспресс-анализа параметров нефти, мазута и битумов [7–11].
Для реализации в автономном ручном режиме нами разработан (рис. 1) и изготавливается портативный переносной, с питанием от аккумулятора «Релаксометр ЯМР NP-1,2».
Анализатор защищен патентом [12], получил золотую медаль на Московском салоне инноваций и инвестиций 2007 г. и Национальный сертификат качества РАЕ. По автономности, минимуму потребляемой мощности и малогабаритности прибор не имеет аналогов и обладает преимуществами по сравнению с лучшими зарубежными аналогами – Minispec Pc120 (Bruker, ФРГ) и MQA 6005 (Oxford, Англия). Отметим, что последние – лабораторные, а не портативные анализаторы.
Опишем один из разработанных нами методов [9].
Определение А, С и П методом ЯМР с одновременным облучением образцов
Каждый УВ имеет определенную полосу поглощения электромагнитного излучения, которое приводит к возбуждению деформационных и валентных колебаний молекул. А это, в свою очередь, ведет к увеличению времен спин-спиновой ядерной магнитной (ЯМР) релаксации протонных фаз Т2А и Т2В, поскольку они пропорциональны частоте и интенсивности молекулярного движения молекул. Селективно возбуждая облучением на требуемой длине волны (нагревая определенные колебательные «моды»), мы можем активировать определенные (парафиновые) молекулы. В результате их времена релаксации T2i принимают завышенные значения по сравнению с обычными, поскольку T2i зависит от τВ, времени корреляции вращательного движения и межпротонного расстояния rij по формуле:
T2i–1 = 3γ4h2τВ/4π2rij6 (1)
где γ = 2,675×104 рад./сек×э – гиромагнитное отношение протонов. Каждой i-й протонной фазе должно соответствовать свое время релаксации T2i. Любое изменение τВ и rij, вызванное облучением, ведет к увеличению интенсивности молекулярного движения – время корреляции τВ падает, а межпротонное расстояние rij из-за увеличения амплитуды колебаний растет. И то и другое ведет к росту T2i возбужденной i-й молекулы.
Запатентованный нами [5] способ оперативного контроля асфальтенов, смол и парафинов включает возбуждение в образце, помещенном в постоянное магнитное поле, сигналов спин-эхо ядерного магнитного резонанса (ЯМР) сериями радиочастотных импульсов, регистрацию амплитуд спин-эхо в измеряемом образце, измерение времен релаксации Т2i без облучения и Т2i* при непрерывном облучении образца светом в видимом и БИК диапазоне спектра, вычисляют относительные изменения Т2i по формуле
ΔТ2i = (Т2i* –Т2i)/Т2i (2)
и определяют концентрации Ci i-й компоненты или соединения по формуле:
Ci = k1i + k2i (ΔТ2i)k3i. (3)
Значения коэффициентов k1i – k3i зависят от вида определяемой i-й компоненты или соединения. Устройство для реализации способа состоит из разработанного нами релаксометра ЯМР и источника излучения по патенту [12].
В парафинах при использовании источника сλ = 1,85 мкм произойдет закачка энергии ΔЕ облучения и парафиновая молекула получит энергию ΔЕ = 1,2×105 /1850 = 64,86 кДж/моль = 0,67 эВ, достаточную для возбуждения вращательных и колебательных движений. Так, измеряя изменение времен ΔТ2В (именно в протонную фазу В дают вклад колебания молекул парафинов) можно оценить их количество в образце.
Изменение концентрации парафина ведет к крутому росту ΔТ2В (рис. 2), который можно оценить с коэффициентом корреляции R2 = 0,988 из уравнения
П = 0,02 (ΔТ2В)3. (4)

Рис. 1. Портативный переносной, с питанием от аккумулятора «Релаксометр ЯМР NP-1,2»
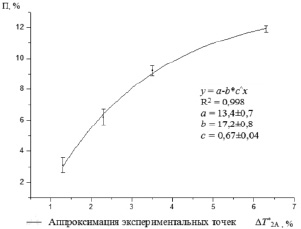
Рис. 2. Корреляция между концентрацией парафинов (П) и увеличением времен релаксации ΔТ2В фазы В в результате облучения на длине волны λ= 1,825 мкм
Абсолютные погрешности по ГОСТу и методу ЯМР с облучением
|
Компоненты |
Парафины |
Асфальтены |
Смолы |
Время измерения (мин) |
|
Абс.погрешность по ГОСТ 8.563-96 |
± 0,3 % |
± 0,6 % |
± 0,1 % |
480 |
|
Абс. погрешность по ЯМР с облучением |
± 0,2 % |
± 0,37 % |
± 0,47 % |
2–3 |
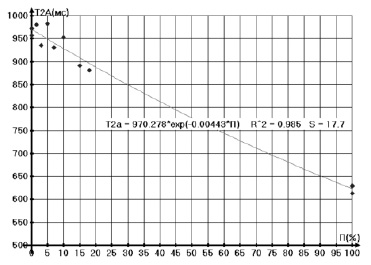
Рис. 3. Зависимость времен спин-спиновой релаксации Т2А от концентрации парафина П
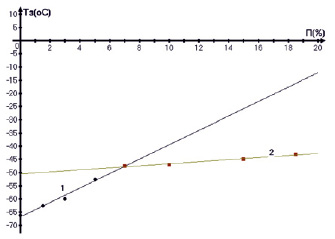
Рис. 4. Зависимость температуры застывания ТЗ дизельного топлива от концентрации парафина П и времени спин-спиновой релаксации Т2А. Точки о – ТЗ(П), ● – ТЗ(Т2А)
Для реализации способа использовалась нефть Ромашкинского месторождения скв. 109, плотностью ρ = 867,5 кг/м3, в которой исходно содержались парафины в количестве 3,1 %, которые дали прирост значений ΔТ2В на величину в 1,3 %. Добавка в нефть парафина в количестве 0,5; 1 и 2 г н-декана С10Н22 дало содержание парафина в образцах – 6,2; 9,2 и 11,9 % вес. в объеме 15 мл. Анализ парафиновых соединений осуществляли устройством, состоящим из релаксометра ЯМР на частоту 9 МГц и некогерентного перестраиваемого излучателя с лампой ТРШ 1500-2300 (ТУ 16.535.847-74). Использован светофильтр на λ = 1,85 мкм. Время измерения составило 2 минуты. Основные погрешности измерений концентраций П, А и С характеризуются значениями, представленными в таблице.
Кроме того, были исследованы зависимости времен Т2Аи Т2В протонных фаз А и В и населенностей протонов РВ фазы В от концентрации парафина в дизельном топливе, измеренные соответственно при температурах 50 и 16 °С и представленные на рис. 3, 4.
Определение концентрации парафинов П и температуры застывания ТЗ, например, в дизельном топливе, осуществляется по зависимости времен спин-спиновой релаксации Т2А протонной фазы А от концентрации парафина (рис. 3).
Можно предполагать, что по мере увеличения концентрации парафинов П должна возрастать их степень ассоциации и, следовательно, наблюдаться укорочение времен релаксации Т2В протонной фазы В и рост населенностей протонов РВ (точки ● и ●), относящихся к парафинам на рис. 3. В то же время должно наблюдаться обеднение парафинами нафтеновой фазы ДТ, т.е. должен быть рост Т2А. Именно это мы и наблюдаем на экспериментальных зависимостях, за исключением того, что рост Т2А имеет место только до 6–8 % П. Видимо далее высокая концентрация П ограничивает молекулярные движения углеводородных цепей и укорачивает оба времени релаксации. В соответствии с предложенной моделью наблюдаются и зависимости ТЗ(П), ТЗ(Т2А) (точки о и •) температуры застывания от концентрации парафина и Т2А с изломом ТЗ(П) при 7 % П.
Библиографическая ссылка
Кашаев Р.С. ЭКСПРЕСС-АНАЛИЗ КОНЦЕНТРАЦИИ ПАРАФИНА И ТЕМПЕРАТУРЫ ЗАСТЫВАНИЯ ТОПЛИВА МЕТОДОМ ЯМР // Современные наукоемкие технологии. 2015. № 8. С. 31-35;URL: https://top-technologies.ru/en/article/view?id=35094 (дата обращения: 02.07.2026).