


Scientific journal
Modern high technologies
ISSN 1812-7320
"Перечень" ВАК
ИФ РИНЦ = 1,279
REACTIONS OF OXIDATION OF ANABAZIN AND HIS DERIVATIVES

В настоящее время синтетические трансформации природных соединений прочно вошли в число ведущих направлений фармацевтической науки. Это связано с уникальной структурой и биологическими свойствами растительных веществ, синтезируемых в результате сложных биохимических процессов жизнедеятельности растений. Изменяя структуру природных веществ можно получить новые, порой уникальные лекарственные препараты, которые оказываются в десятки раз более эффективными и менее токсичными биоактивными веществами, чем исходные субстраты.
Одним из перспективных в плане модификации строения соединений является известный алкалоид анабазин, широко использовавшийся в 1930-1960 гг. как мощное инсектицидное средство против различных вредителей сельскохозяйственных культур. Анабазин был первым алкалоидом, выделенным из растительного сырья Anabasis aphylla в бывшем Советском Союзе в 1922г. Ядовитые и лекарственные свойства анабазина давно были известны местному населению. Так, например, порошком, полученном из стебля, присыпали раны; отваром корней лечили туберкулез. Отваром растения пользовались для уничтожения личинок, заводящихся в ранах животных. В начале 60-х годов в связи с появлением высокоэффективных и быстродействующих фосфорорганических инсектицидов, анабазин сульфат был снят с производства [1, 8].
Анабазин (C10H13N2) является структурным изомером известного алкалоида никотина, сходен с ним не только по химической природе, но и по физиологическим свойствам. Строение анабазина соответствует a-пиперидинил-b-пиридину, т.е. характеризуется наличием в молекуле пиперидинового фрагмента со вторичной аминофункцией и пиридинового фрагмента с характерным для него ароматической делокализацией электронной плотности, которые в целом и определяют особенности реакционной способности алкалоида анабазина (7).
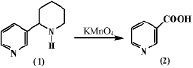
Процесс окисления является одним из интересных реакций изучения химической модификации анабазина. В 1931 году Орехов А.П. и Меньшиков Г.П. [9] окислили анабазин с применением марганцовокислого калия и получили никотиновую кислоту (2), являющуюся основным компонентом витамина РР.
Для нахождения оптимального способа синтеза никотиновой кислоты Матвеев В.В. [4] окислял анабазин концентрированной серной кислотой и кислородом воздуха в присутствии различных катализаторов (металлический селен, ртуть, сернокислый висмут, осмиевая кислота, пятиокись ванадия). Результаты исследований показали, что в изученных условиях лучшим катализатором является металлический селен. Наибольший выход никотиновой кислоты при этом составил 77,5 %.
В работе [9] приведены данные по синтезу никотиновой кислоты из доступного технического анабазина (смесь сульфатов анабазина и лупинина). Показано, что окисление смеси анабазина и лупинина азотной кислотой имеет ряд преимуществ перед окислением анабазина перманганатом, хотя при этом образуются нитраты никотиновой и лупининовой кислот, но разделение их не представляет особых затруднений благодаря их различной растворимости в воде. Выход никотиновой кислоты от исходной смеси составляет около 74 %, или около 26 % от технического анабазин-сульфата.
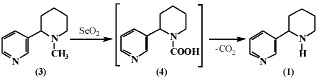
Также в работе [9] приведены данные по окислению анабазина (1) и N-метиланабазина (3) с применением селенистого ангидрида. При окислении анабазина селенистым ангидридом процесс не протекает, несмотря на проведение процесса в жестких условиях. N-метиланабазин (3) в этих условиях окисляется до анабазина (1) через образование неустойчивого промежуточного продукта – N-карбоновой кислоты (4), которая легко расщепляется с выделением углекислого газа. Также было установлено, что ядра пирролидина и пиперидина устойчивы по отношению к селенистому ангидриду.
Следует также отметить, что N-метиланабазин подвергается электрохимическому окислению с образованием никотиновой кислоты (выход 15 %).
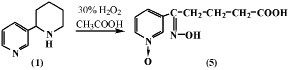
Известно, что перекись водорода, а также ее комплексное соединение с мочевиной гидропирит, способны окислять алкилбензолы до соответствующих алкилбензойных кислот. В работе [2] описано окисление анабазина (1) действием 30 %-й Н2О2 в уксусной кислоте. Показано, что реакция протекает окислением и пиридиновой и пиперидиновой частей алкалоида анабазина с образованием продукта – Ру-N-окиси δ-оксимино-δ-(пиридил-3)валериановой кислоты (5).
При действии 10 %-й Н2О2 в водном растворе окисляется только пиперидиновое кольцо анабазина с образованием δ-оксимино-δ-(пиридил-3)валериановой кислоты (6).
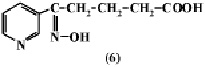
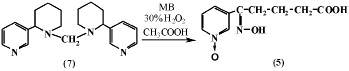
Нами [10-12] изучена реакция окисления анабазина 30 %-ным раствором перекиси водорода в среде ледяной уксусной кислоты в условиях МВ-облучения. Установлено, что продуктом окисления анабазина (1) в изученных условиях является Ру-N-окись δ-оксимино-δ-(пиридил-3)-валериановой кислоты (5). Реакция проводилась в различных условиях с варьированием мощности и времени облучения. Установлено, что при оптимальных значениях – мощность облучения 70 Вт, время облучения от 20 до 30 минут, достигается наибольший выход конечного продукта (65 %), сопоставимый с выходом в классических условиях. Явным преимуществом проведения реакции окисления анабазина в условиях микроволновой активации является значительное сокращение времени реакции – с 24 часов по классической методике [11] до 30 минут в условиях микроволновой активации.
Нами установлено, что соединение (5) образуется также при окислении дианабазинилметана (7) 30 % раствором перекиси водорода в среде ледяной уксусной кислоты при микроволновой активации реакционной смеси (мощность излучения 70 Вт) [9, 10]. Видимо, действие перекиси водорода в условиях МВ-облучения приводит к разрушению метиленового мостика в (7) с последующим окислением молекулы анабазина с образованием Ру-N-окиси δ-оксимино-δ-(пиридил-3)-валериановой кислоты (5).
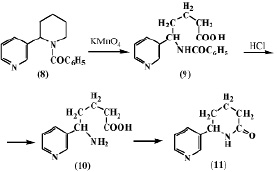
Авторы работы [5] подвергли окислению N-бензоиланабазин (8) марганцовокислым калием для получения анабазона (11). В результате им удалось выделить неактивную d-бензоиламино-d-(b’-пиридил)валерьяновую кислоту (9). Нагревание ее с соляной кислотой приводит к отщеплению бензоильной группы и образованию неустойчивой d-амино-d-(b’-пиридил)валерьяновой кислоты (10), которая легко переходит в лактам a-(b’-пиридил)-a-пиперидон (11).
В работе [9] описан синтез Py-N-окиси анабазина (13) действием перекиси водорода в уксусной кислоте на N-бензоиланабазин (8) с последующим элиминированием ацильного остатка.
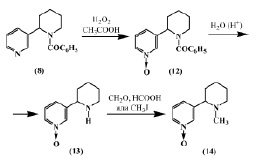
Синтезированная Py-N-окись N-бензоиланабазина (12) гидролизовалась до Py-N-окиси анабазина (13), при метилировании которой формальдегидом и муравьиной кислотой или йодистым метилом образуется Py-N-окись N-метиланабазина (14). Из этих данных следует, что наличие заместителя у атома азота пиперидинового цикла препятствует расщеплению пиперидинового кольца.
Меньшиков Г.П. и др. [6] окисляли йодметилат N-бензоиланабазина (15) железосинеродистым калием и получили соответствующее производное N-метиланабазона (16), при омылении которого образуется N-метиланабазон (17). Соединение (17) при взаимодействии с пятихлористым фосфором отщепляет хлористый метил и образует 2-хлоранабазин (18). Последний при окислении марганцовокислым калием образует 2-хлорникотиновую кислоту (19).
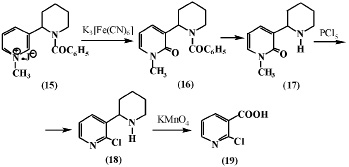
В дальнейшем авторы [6] при изучении окисления йодметилата N-метиланабазина (20) в вышеуказанных условиях получили N,N’-диметиланабазон (21) и N-метил-a-хлоранабазин (22), последний с метилатом натрия образует N-метил-a-метоксианабазин (23).
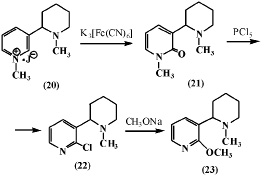
Из данных, представленных в работе [6] следует, что при окислении соединений (15) и (20), имеющих заместитель у атома азота в пиперидиновом фрагменте, его окисления также не происходит.
Таким образом, анализируя данные по окислению анабазина и его производных различными окислителями и в разных средах, можно сделать вывод о том, что процесс окисления протекает с сохранением пиридинового фрагмента, что обусловлено высоким значением энергии делокализации пиридинового цикла. Пиперидиновый фрагмент, входящий в структуру анабазина и его производных, подвергается окислению, так как неподеленная пара электронов атома азота в нем, в отличие от неподеленной пары электронов атома азота пиридинового фрагмента, не участвует в ароматической делокализации электронной плотности. В то же время, присутствие заместителей у атома азота в пиперидиновом фрагменте может привести к стабилизации (дезактивации) его неподеленной пары электронов как за счет электронных, так и стерических факторов в зависимости от природы заместителей.
Библиографическая ссылка
Бакирова Р.Е., Фазылов С.Д., Mуравлева Л.Е., Нуркенов О.А., Животова Т.С., Сатпаева Ж.Б., Жакупова А.Н., Аринова А.Е. РЕАКЦИИ ОКИСЛЕНИЯ АНАБАЗИНА И ЕГО ПРОИЗВОДНЫХ // Современные наукоемкие технологии. 2014. № 9. С. 59-63;URL: https://top-technologies.ru/en/article/view?id=34706 (дата обращения: 03.07.2026).